Dr Emanuele Paci
- Position
- Visiting member
- Areas of expertise
- computational biology; biological physics; computational chemistry
- [email protected]
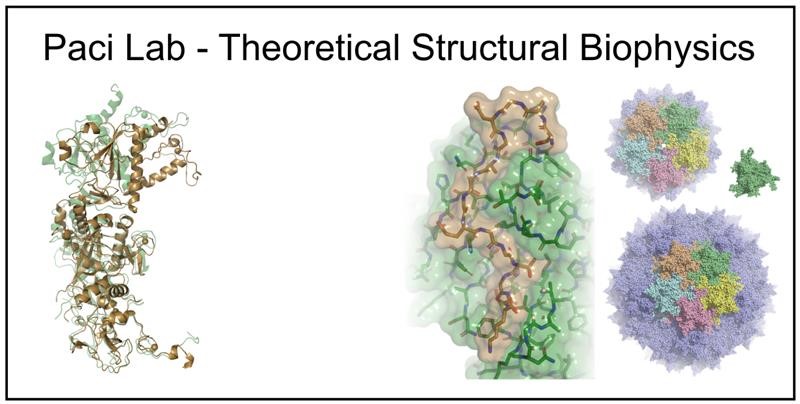
Introduction
Our research focuses on the development and application of computational tools to investigate structure and functional dynamics of biomolecules. One of our aims is fully exploit the wealth of experimental data that is increasingly available thanks to the development of high throughput techniques. Most of our research involves experiments performed in collaboration with colleagues from the Astbury Centre and further afield. A central topic of our research is to develop and use data-driven approaches to determine the relation between sequence and physical properties of biological polymers, and proteins in particular. Data can be generated by computer simulations (e.g., a sample of conformations representing the thermodynamic state of the system) or experimentally measured properties (which range from cytotoxicity or number of hydrogen atoms exchanged with deuterium one in a given time). Much of our research involves molecular simulation of the dynamics and interactions within and between protein systems. Data generated from simulation are used to interpret and direct experimental measurements such as MS, NMR and SAXS.
Current major projects
- Re-engineering of viral capsids for gene therapy
- Disordered polypeptides and effect of phosphorylation on conformational propensity
- Sugar-binding proteins, adhesion and design of inhibitors
- Hydrogen-deuterium exchange
- Detailed Research programme
Detailed research programme
Re-engineering of viral capsids for gene therapy
Viral capsids are amazing molecular entities where complex functional structures form by self-assembly of multiple identical proteins. This presents an interesting opportunity for exploiting computational predictive methods to redesign such assemblies, for example with different stoichiometry, by exploiting symmetries and multivalency, or modify the sequences to improve their properties. In collaboration with companies developing gene therapeutics, we focus on developability issues (e.g., increase the genome capacity, improve the stability capsids, preventing their aggregation) that can be addressed through a small number of mutations.
Disordered polypeptides and effect of phosphorylation on conformational propensity
Short peptides are particularly amenable to study by molecular simulation. After discovering the physical principles behind propensity to form helices of some polyampholyte peptide sequences, and how the alteration of specific charge patterns in the sequence causes the peptides to become disordered we are investigating, in collaboration with Richard Bayliss (Astbury) the effects of phosphorylation on polypeptide structure, specifically where phosphorylation of serine can enhance helicity in peptides and bound protein complexes when supported by interaction with neighbouring basic residues. Phosphorylation is a key process in cell signalling so understanding the often-overlooked effects on structure could yield useful insights, for instance, in inhibitor design.
Hydrogen deuterium-exchange
The exchange of hydrogen atoms of a protein in deuterated solvent has been used as a probe for structure and dynamics of proteins for decades, and provided key insights on the mechanism of protein folding. Recent advances in mass spectrometry allow fast measurement of the exchange kinetics, with small, unlabelled samples, for random fragments of the protein. Using theoretical models and statistical learning we developed a method than amplifies the resolution to single residue level. We are now, in collaboration with Frank Sobott, extending the method, and integrating other MS measurements to provide detailed structural and dynamical representations of proteins, including their disordered and aggregated states.