Professor Richard Bayliss
- Position
- Professor of Molecular Medicine
- Areas of expertise
- Dynamic protein-protein interactions; Protein kinases; Cancer signalling; Structure-based drug design; Cell division
- [email protected]
- Phone
- +44(0)113 343 9919
- Location
- 6.108a Astbury
- School
- Molecular and Cellular Biology
Introduction
My team studies the molecular structures and interactions of proteins using structural, cell, chemical and computational biology approaches. We work towards understanding the molecular mechanisms that underpin cellular functions and how these events are altered in human diseases such as cancer. In partnership with other scientists, we develop precision drugs that are tailored to individual proteins.
Current major projects
- Dynamic protein interactions
- Protein kinases and cancer drug discovery
- Cancer signalling pathways
- Cell Division
Detailed research programme
Dynamic protein interactions
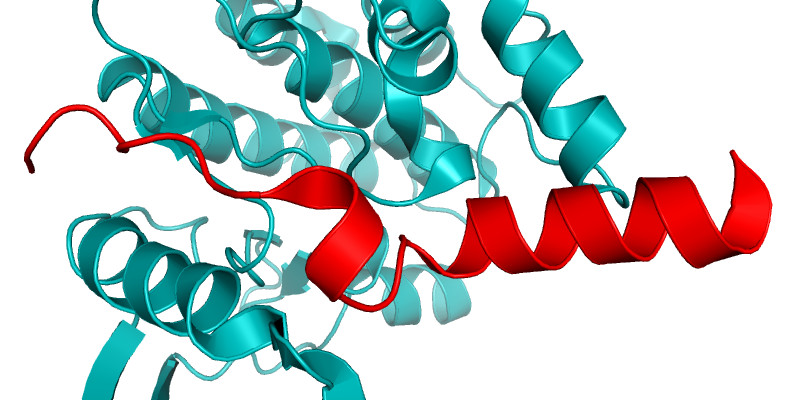
Dynamic cellular processes such as the assembly and function of the mitotic spindle, and the transport of macromolecules between the nucleus and cytoplasm, are the product of dynamic interactions between protein molecules. These interactions may involve regions of proteins that are intrinsically disordered, and which adopt a regular structure only transiently whilst in complex with their partner. We investigate these fascinating systems using a combination of structural, computational and cell biology approaches. One example is the MYC family of transcription factors whose interactions are regulated by Aurora-A (Büchel Cell Reports 2018). The crystal structure of the Aurora-A/N-MYC complex explained why this interaction stabilizes N-MYC and how this can be reversed by certain inhibitors of Aurora-A (Richards, PNAS 2016).
Protein kinases and cancer drug discovery
Protein kinases are frequently mutated or otherwise dysregulated in cancer and inhibitors of protein kinases such as imatinib and crizotinib are key therapeutic drugs. Our research aims to determine the structural mechanisms that underpin kinase regulation and to develop new kinase inhibitors as potential cancer therapeutics. We resolved the allosteric mechanism by which the non-catalytic, C-terminal domain (CTD) of NEK9 activates NEK7 (Haq, Nat. Comm. 2015) and showed that IRE1 is regulated through a similar mechanism (Joshi, Oncotarget 2015). We have assisted in the development of inhibitors of the protein kinases Aurora-A, NEK2 and IRE1 and have ongoing collaborations on several other targets.
 Cancer signalling pathways
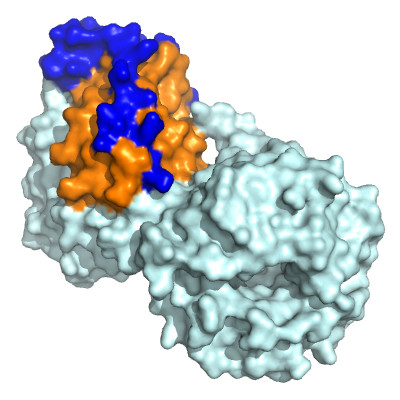
Cancer signalling pathways
We are investigating the signalling processes that underpin cancer cell proliferation and survival. The EML4-ALK fusion oncoprotein is responsible for ~5% of non small cell lung cancer. We determined the structure of the trimerization domain of EML4, which is essential for oncogenic ALK signaling (Richards, Biochem J. 2015) and the structure of the TAPE domain of the related protein EML1 (Richards, PNAS 2014). Working with collaborators in Seoul, the structures helped to stratify EML4-ALK patients with variant forms of the fusion, revealing that a subset of patients respond poorly to therapy (Woo, Ann Oncol 2017; Sabir Cancers 2017). The EML4-ALK variant in these patients promotes changes in microtubule organisation and cell migration through the NEK7 and NEK9 kinases (O’Regan, 2020).
 Cell Division
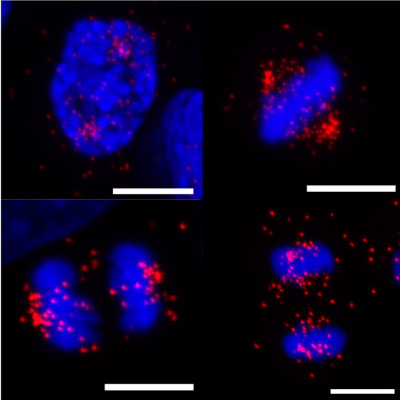
Cell Division
Microtubules are dynamic structures that form part of the cellular cytoskeleton. During mitosis, microtubules form the bipolar spindle structure that carries out chromosome division. Errors in this process lead to aneuploidy, chromosomal instability and supernumerary centrosomes, which are common features of tumour cells and arguably drivers of cancer. Our main project in this area aims to resolve the intermicrotubule bridge structures that reinforce the mitotic spindle. The shortest bridges are formed by the clathrin/TACC3/ch-TOG (CTC) complex, assembly of which is controlled by the protein kinase Aurora-A. The spindle protein TACC3 is a dynamic, disordered protein, which is phosphorylated by Aurora-A, and adopts a helical conformation as it binds its partner CHC (Burgess, EMBO J. 2018).