Professor John Ladbury
- Position
- Professor of Mechanistic Biology
- Areas of expertise
- Intracellular Signal Transduction, Cancer Molecular Mechanisms, Molecular Biophysics Drug Development, Protein-Protein Interactions
- [email protected]
- Location
- LIGHT Building
- Faculty
- Biological Sciences
- School
- Molecular and Cellular Biology
- Website
- ORCID
Introduction
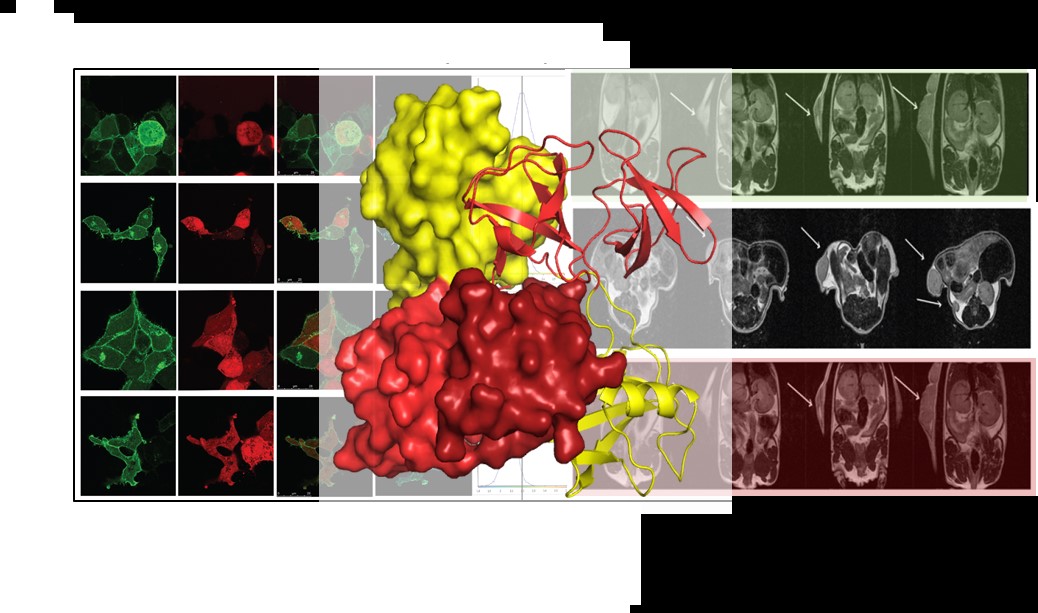
Receptor tyrosine kinases (RTKs) expressed on the plasma membrane of cells in normal tissue are rarely exposed to high concentrations of extracellular growth factors. Nonetheless they express proteins associated with kinase-mediated signalling. We are interested in the signalling associated with these protein that occurs under basal conditions (which are close to the conditions experienced by normal tissue. In the absence of extracellular stimulation or genetic mutation, an oncogenic response can be driven by the competitive binding of SH3 domain-containing downstream effector proteins to proline-rich sequences on growth factor receptors. Of the approximately 50 plasma membrane receptor tyrosine kinases (RTKs) the majority have proline-rich sequences in their C-termini. These have a propensity to bind to the >300 proteins expressed in human cells which contain SH3 domains. These interactions occur in the absence of any extracellular stimulation (e.g. growth factors, cytokines). Proline-rich sequence binding to SH3 domains are promiscuous and the observed interactions with RTKs are dependent on the relative concentrations of the proteins involved. This form of non-canonical signalling represents a second tier (Tier 2) of RTK-mediated response, different from normal kinase activated pathways. We have demonstrated that Tier 2 is important in cellular homeostasis and metabolic control, however under conditions of cellular stress can give rise to cancer and other pathologies.
Current major projects
- Proteomic screening cancer cells for RTK proline-rich sequences and their binding partners
- Validation of key Tier 2 signalling pathways using biophysical and structure methods
- Investigation of the role of phase separation of signalling components on downstream RTK-mediated response
- Validation of signalling in C. elegans organism models
Detailed research programme
We previously established that under non-stimulatory conditions the fibroblast growth factor receptor 2 (FGFR2) recruits the adaptor protein, growth factor receptor binding protein 2 (Grb2) through its C-terminal SH3 domain. In cells depleted of Grb2 other proteins can access the proline-rich motif on FGFR2. One of these proteins, phospholipase C(gamma)1 (Plcγ1) is activated on binding and through turnover of plasma membrane phospholipids to produce second messengers, raises cellular calcium levels which are responsible for increased cell motility and invasive behaviour. In ovarian and lung adenocarcinoma patients with low levels of Grb2 and increased expression of Plcγ1 higher incidence of metastasis leads to greatly reduced survival outcomes.
The dependence of signalling described above on respective concentrations of RTKs and SH3 domain containing proteins mean that there is no on-off switch for this form of signalling, the outcomes are dictated solely by fluctuations of protein concentrations. As a result one key driver for this form of signalling is cellular stress. We are working to establish how stresses experienced by tissue (e.g. pH change associated with acid reflux in the oesophagus) can lead to cancer outcomes.
We have extended our studies in this area to explore other RTK-SH3 domain-containing protein interactions to establish whether the up-regulation of signal transduction through these interactions is a general phenomenon. This leads to the hypothesis that two tiers of intracellular signalling can be derived from receptors with intrinsic protein kinase activity:
1) Ligand-induced elevation in kinase activity resulting in tyrosylphosphate-mediated effector protein recruitment and committal to a defined cellular outcome (e.g. proliferation).
2) Receptor phosphorylation-independent activation of downstream effectors through SH3 domain/proline-rich sequence interactions, which appear to be required for cell homeostasis/metabolic control.
Hyperactivity of the Tier 1 signalling is a feature of receptor tyrosine kinase-related cancers arising from genetic mutation. Although the tier 2 signalling mechanism occurs under basal conditions, and is thus likely to be associated with cellular maintenance, we have shown that fluctuations in expression levels of SH3-containing proteins can drive cells into pathological phenotypes including proliferation and metastasis.
We are testing this hypothesis with a range of methods extending from cell-based assays (including fluorescence lifetime imaging microscopy) through to structural and in vitro biophysical analysis.
We are optimising a screening protocol to establish the extent of Tier 2 signalling in a range of cells and conditions. We have identified novel interactions involving well studied proteins as well as less understood systems. These are being validated and phenotypic outcomes of knocking down these interactions are being explored to establish the effects of signalling in normal tissue.
We have focused on gastro-intestinal cancers we have begun to explore the effects of stress on intracellular protein expression and the outcomes on Tier 2 signalling. We have shown that by mimicking conditions experienced in the GI tract we can affect expression of receptor tyrosine kinases.
In addition to identifying the signalling pathways which are initiated as a result of fluctuations in protein concentrations in cell-based assays, we are exploring the interactions associated with up-regulation of Tier 2 signalling using both biophysical and structural biological methods. High resolution structural detail on the receptor-ligand interactions are providing invaluable detail on the mode of recruitment of signalling proteins as well as information towards potential inhibition of aberrant pathways that lead to pathogenic outcome.